Application Note CloneSelect Imager FLによる
遺伝子編集セルラインの高速化
- トランスフェクションに成功した細胞を容易に検出可能
- 細胞株とコンフルエンスの追跡時間を短縮
- トランスフェクション効率の低いプールを早期に排除
- マルチチャンネル蛍光検出によるCRISPR編集の確認と追跡 蛍光検出
- オーバーパスエイジングのリスクを低減
PDF版(英語)
はじめに
Prathyushakrishna Macha, PhD | Research Scientist | Molecular Devices
遺伝子編集とCRISPR技術
遺伝子編集技術は、DNAレベルでの操作を可能にするもので、さまざまなツールを用いて塩基配列の欠失、付加、改変を行う。近年、あらゆる生物のゲノムを編集するための効率的な戦略が数多く考案され、新たな手法もかつてないほど進歩している。遺伝子組み換えは、基礎研究、病気との闘い、バイオ製品の製造など、幅広い応用範囲をカバーしている。これらの用途には、疾患モデルの開発、医薬品開発、標的遺伝子の機能解析、トランスジェニック生物の作製、キメラタンパク質の生産などの特異性がある。ゲノム編集に利用できるプログラム可能なヌクレアーゼには、ジンクフィンガーヌクレアーゼ(ZFN)、メガヌクレアーゼ、転写活性化因子様エフェクターヌクレアーゼ(TALEN)、CRISPR(Clusteredly Regular Interspaced Short Palindromic Repeats)関連タンパク質9(Cas9)などがある。標的遺伝子編集にはこれらの既存技術が用いられるが、特にCRISPRは、優れた効率性、使いやすさ、多重化可能性を示すことで科学に革命をもたらした1,2。次世代CRISPR-Cas技術は、DNAだけでなくRNAも標的とすることを目指しており、遺伝子治療や細胞治療における新たな最先端ツールが期待されている。
P53遺伝子の意義
ヒトのp53は、20kb、11のエクソンと、最初の2つのエクソンを隔てる10kbのイントロンを持つ遺伝子で、ヒトの発癌と腫瘍抑制に重要な役割を果たすとして、常に関心を集めてきた。この遺伝子は、細胞内の傷ついたDNAが複製されないようにする成長抑制作用を持っている。この遺伝子はまた、アポトーシス活性によって制御され、損傷を受けたセルを体内から除去し、正常な細胞の増殖を促進する。ヒトの癌の約50%がp53遺伝子の変化を報告しており、p53の制御を理解することは、薬剤/低分子化合物設計の基礎となる。これにより、癌におけるp53再活性化療法が可能になり、p53に基づく化学療法抵抗性と闘うことができる3-8。
細胞株開発の課題と我々のアプローチ
CRISPR/ Cas9やプライム編集などの遺伝子編集技術の台頭は、基礎研究や疾患研究のための細胞モデル作製の加速化に貢献している。遺伝子編集後の細胞のスクリーニングは、一般的に手作業で行われる重要なステップである。これは、膨大な時間と手作業を伴う面倒なプロセスである。効果的な遺伝子編集プロセス(より高いトランスフェクション効率)のためには、核酸ベクターのデザイン(ターゲットとのコンストラクト)、宿主細胞株の特徴、デリバリー方法のような複数のパラメーターを最適化しなければならない。これを手作業で大規模に行うのは大変なことである。遺伝子編集、安定細胞株スクリーニング、モノクローナル細胞株開発の再現性とスケーラビリティを高めるには、効率的なワークフローが不可欠です。
CloneSelect® Imaager FLは、コンフルエンス追跡と蛍光イメージングチャンネルを使用して、最大96または384のトランスフェクション条件のスクリーニングを可能にし、レポーター遺伝子の発現検出を通してトランスフェクションをうまくモニターします。マルチチャンネル蛍光イメージャーにより、効率が大幅に改善され、ワークフローが合理化され、細胞プールが生成され、最終的にはデイゼロのモノクローナル性追跡が可能なモノクローナル細胞株が生成されます。CloneSelect Imager FL システムは、蛍光チャンネ ルを使用して増殖(懸濁または接着)およびトランスフェクション した細胞を効率的にイメージングします。これらの画像を解析することで、最も高いトランスフェクション効率と目的のタンパク質発現レベルを選択することができます。CloneSelect Imager FL システムには、2つの蛍光チャンネルと、コンフルエンス検出用の白色光イメージングがあります。
CRISPRによる遺伝子編集のSTEPSとその検証
ここでは、CRISPR編集プールを作製するために、宿主細胞株としてHEK-293を選択し、p53ノックアウト(KO)系を選択した。p53は、成長停止やストレスに対するアポトーシス関連応答をアップレギュレートすることが知られており、その結果、プログラムされた細胞死、細胞周期、分化の変化を引き起こす。編集されたセルは、p53ノックアウト確認のために抗体でタグ付けされ、次いでアポトーシスアッセイによって、改変型と野生型のHEK-293細胞におけるp53ノックアウトの効果を比較した。
方法
細胞培養とトランスフェクション
ATCCのHEK-293細胞は、Santa Cruz p53 CRISPR/Cas9 KOプラスミドとp53 Homology-directed repair (HDR)プラスミドを用いてTP53ノックアウトを行う前に、製造者の指示に従って維持した(Santa Cruz Biotechnology)。細胞は96(Corning 3300)ウェルプレートに50k/ウェル播種密度で播種し、トランスフェクション当日は、TransIT-X2デリバリーシステム(Mirus Bio)に付属の説明書に従い、無血清培地中で細胞は約60%コンフルエントになった。CRISPR/Cas9 TP53 KOプラスミド(sc-416469)とHDRプラスミド(sc-416469-HDR;Santa Cruz Biotechnology)を異なる比率(1:2、1:3)で選抜用試薬と共トランスフェクトし、トランスフェクション後24時間の細胞をピューロマイシンで選択した。
CloneSelect Imafer FL を用いた抗生物質選択。
ピューロマイシンの濃度は、最初の72時間は1.5 µg/mLで、その後、培地交換により徐々に0.5 µg/mLに漸減させ、安定した株形成まで定期的に維持した。CloneSelect Imager FLマルチチャンネル・コンフルエンス・ランで、ピューロマイシン選択前と選択後の細胞を定期的にモニターした。細胞は追跡され、培地は2~3日ごとに交換され、細胞は48ウェル組織培養処理プレートに移され、次のアッセイのために6ウェルプレートに移された。
免疫染色
増殖後、細胞を組織培養処理した96ウェルプレートでコンフルエントになるまで増殖させ、4%パラホルムアルデヒドで固定し、PBSを3回交換して洗浄した。これらの細胞を、Alexa Flour 647 (sc-126 AF647, Santa Cruz Biotechnology)、Phalloidin 488(1:400)、Hoechst 3342(1:1000)に3% Blocking reagent (BSA)を結合させたp53 Antibody (DO-1)で一晩標識し、PBSを3回交換して洗浄した。画像は、Molecular Devices社のImageXpress® Micro Confocal High-Content Imaging System (IXM-C)の10X対物レンズを用いて取得した。
アネキシンV
プレーティングから48時間後、HEK-293細胞の96ウェルプレートを1:2連続希釈のスタウロスポリン(5μM)に4時間暴露し、アポトーシスを誘導した。アポトーシスは、アネキシンV-FITCアポトーシスキット(ab14085)を用いて、製造者の指示に従って検出した。画像はIXM-Cの10X対物レンズを用いて取得し、結果はMetaXpress®およびSoftMax® Proソフトウェアを用いて解析した。
結果と考察
CRISPR/Cas9システムを用いてHEK-293のp53遺伝子をノックアウトするために、ターゲティング用と抗生物質選択用の2種類のベクターを選択し、脂質ベースのシステムを用いて一過性にトランスフェクトした。トランスフェクション効率は、DNAとトランスフェクション試薬を1:3の割合でトランスフェクトしたセルで高かった。トランスフェクション試薬を増やすほど効率は高くなったが、潜在的な細胞毒性を避けるため、この比率はそれ以上増やさなかった。p53 KOプラスミドでトランスフェクトに成功した細胞は緑色(GFP)に、p53 KOとHDRの両方を導入した細胞は赤色(RFP)に蛍光を示した(図3 A, B参照)。トランスフェクション効率の迅速な評価とピューロマイシンによるスクリーニングは、CloneSelect Imager FLを用いて行った(図1、2、3)。これらのセルをさらに選別し、p53 KO細胞の安定したプールを得た(図3-上)。
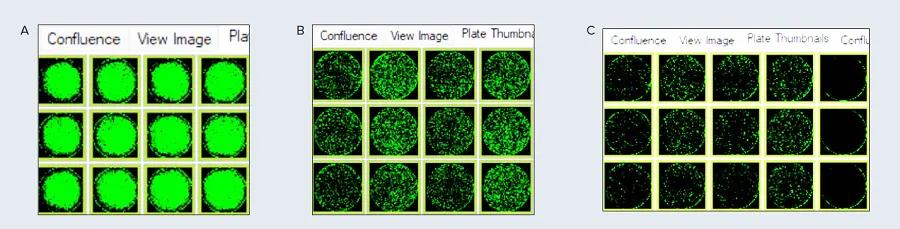
図1. p53ノックアウトのためのCRISPRベースのプラスミドをトランスフェクションした後の、ピューロマイシン選択培地中のHEK-293細胞のプレートサムネイル。CloneSelect Imager FLを用いてレポーターシグナルとコンフルエンス測定を追跡し、さらなる拡大を行った。A. 生細胞の白色光イメージングと全生細胞のコンフルエンシーの表示(ソフトウェアにより擬似緑色ハイライトを適用) B. GFPイメージング、GFPレポーターを有するp53 KOプラスミドでトランスフェクトに成功したことを示す(擬似緑色でコンフルエンシーの表示) C. RFPイメージング、RFPレポーターを有するHDRプラスミドでトランスフェクトに成功したことを示す(擬似緑色でコンフルエンシーの表示)。
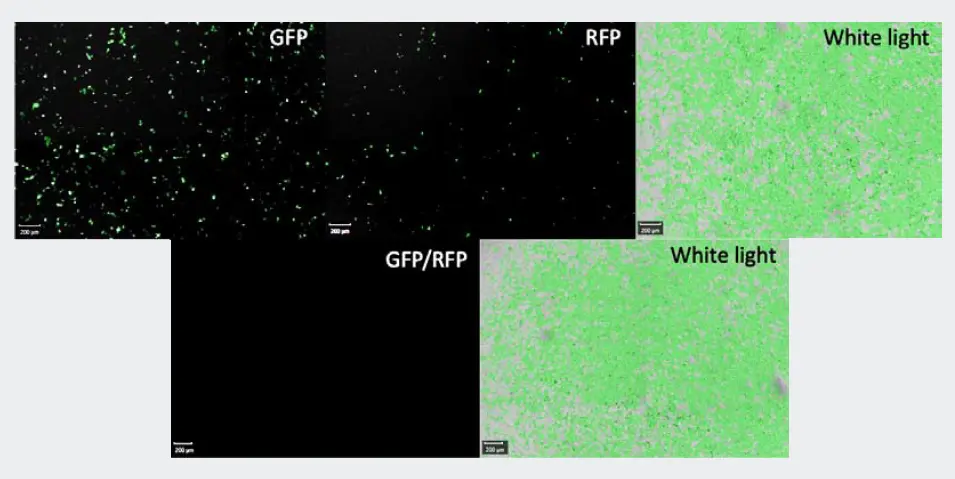
図2. GFP(左)、RFP(中)、白色光フィルター(右)でのA1(上)とA5(下)のウェルイメージ(擬似緑色細胞コンフルエンスジェネレーター)。A1はノックアウトと抗生物質耐性のためにCRISPRベースのプラスミドをトランスフェクトしたのに対し、A5はトランスフェクトしていないため、蛍光タンパク質を用いた細胞分布の報告がない(左下)。
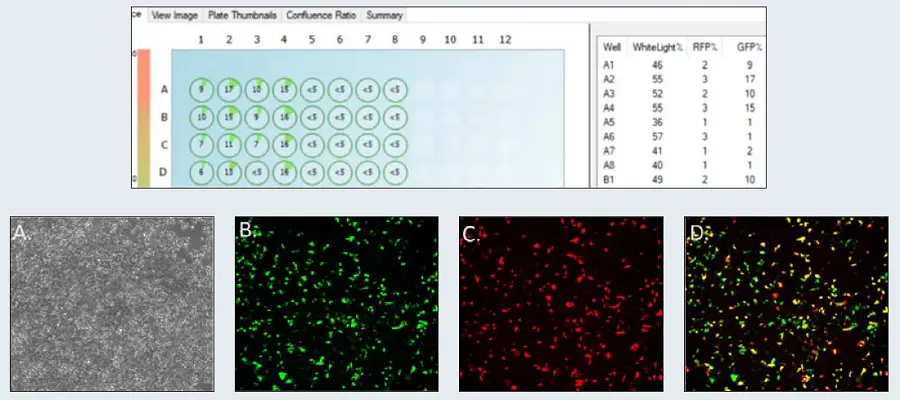
図3. トランスフェクトしたHEK-293細胞とコントロール細胞のピューロマイシン選択時に表示された各細胞のコンフルエンスと細胞数の推定。細胞の分布は、TL、GFP、およびRFPイメージング用のソフトウェアによって推定される(上部の数字パネル)。ピューロマイシン・スクリーニング前のトランスフェクション後の細胞のImageXpress画像(下段の画像パネル)。A. トランスフェクトしたHEK-293(全細胞)のTL画像、B. FITC画像(GFP発現細胞)、C. Texas Red画像(RFP発現細胞)、D. BとCのオーバーレイ(GFPとRFPの両方を発現)。
生成されたプールは、より良い増殖のためにピューロマイシンでさらに選択され、さらなるアッセイのためにスケールアップされた。スクリーニングに成功したHEK-293細胞とコントロールのHEK-293細胞でp53の抗体染色を行ったところ、p53の発現がないことが示された(図4B)。p53のノックアウト成功は、アポトーシスアネキシンVアッセイに基づく二重染色でさらに検証された。KO細胞とコントロールHEK-293細胞のアポトーシス率には有意差があった。p53はアポトーシスにおいて重要な役割を担っているため、p53ノックアウトはスタウロスポリン処理で誘導されるアポトーシス活性を有意に低下させた(図5)。赤のグラフはコントロールのウェル、青のグラフはp53ノックアウトのウェルにそれぞれ対応する(図5)。スタウロスポリンの最高濃度(5μM)では、編集細胞とコントロール細胞の間にほぼ10倍の差があった。薬物濃度が下がるにつれて倍率の差は小さくなったが、それでもコントロール細胞の方が全体的に高いアポトーシス活性を示した。
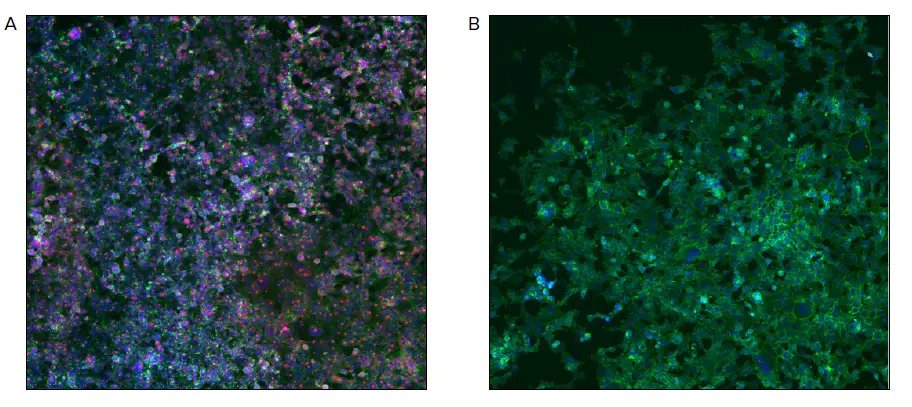
図4. Alexa Flour 647 (sc-126 AF647, Santa Cruz Biotechnology)、Phalloidin 488(1:400)、Hoechst 3342(1:1000)を結合させたp53抗体(DO-1)で標識したp53 KO HEK-293細胞(B)とコントロール細胞(A)の合成イメージング。

図5. Annexin V-FITC(トップ画像パネル)、白色光イメージング、FITCイメージのオーバーレイによって検出されたセルアポトーシス。A. Staurosporine(2.5μM)で処理したp53 KO細胞とB. Staurosporine(2.5μM)で処理したコントロール細胞。ImageXpressで得られたデータをMetaXpress®ソフトウェアで解析したグラフ(下)。処理細胞のアポトーシス活性を示すFITCシグナルをスタウロスポリンの濃度に対してプロットした。
結論
本研究では、CRISPRを用いた遺伝子編集と抗生物質スクリーニングを用いて、p53ノックアウトHEK-293セルプールを開発するための効果的かつ迅速なプロセスを確立した。これは、当社のCloneSelect Imager FLを用いて効果的に実施された。さらに、p53のノックアウトを評価するために、様々なイメージングアッセイを用いて細胞を調べた。このノックアウトにより、アポトーシス誘導時の細胞プールの平均アポトーシス活性が大幅に低下した。CloneSelect Imager FLにより、蛍光検出による遺伝子編集細胞の効率的なスクリーニングが可能になった。
参考文献
- Maeder, M. L., & Gersbach, C. A. (2016). 遺伝子・細胞治療のためのゲノム研究。Molecular Therapy, 24(3), 430-446.
- Khalil, A. M. (2020). ゲノム編集革命。Journal of Genetic Engineering and Biotechnology, 18(1), 1-16.
- Haupt, S., Berger, M., Goldberg, Z., & Haupt, Y. (2003). アポトーシス-p53ネットワーク。Journal of cell science, 116(20), 4077-4085.
- Amaral, J. D., Xavier, J. M., Steer, C. J., & Rodrigues, C. M. (2010). アポトーシスにおけるp53の役割。Discovery medicine, 9(45), 145-152.
- Brown, C. J., Lain, S., Verma, C. S., Fersht, A. R., & Lane, D. P. (2009). 守護天使の目覚め:p53経路の薬物療法。Nature Reviews Cancer, 9(12), 862-873.
- Liu, Y., Wang, X., Wang, G., Yang, Y., Yuan, Y., & Ouyang, L. (2019). がん治療のためのp53-MDM2/MDMXを標的とする潜在的低分子薬の過去、現在、未来。European journal of medicinal chemistry, 176, 92-104.
- Cao, X., Hou, J., An, Q., Assaraf, Y. G., & Wang, X. (2020). p53変異を介した抗がん剤耐性の克服に向けて。Drug Resistance Updates, 49, 100671.
- Stiewe, T., & Haran, T. E. (2018). 変異がp53とゲノムとの相互作用をどのように形成し、腫瘍形成と薬剤耐性を促進するか。Drug Resistance Updates, 38, 27-43.
- Pickar-Oliver, A., & Gersbach, C. A. (2019). 次世代のCRISPR-Cas技術と応用。Nature reviews Molecular cell biology, 20(8), 490-507.
- Gao, P., Lyu, Q., Ghanam, A. R., Lazzarotto, C. R., Newby, G. A., Zhang, W., ... & Miano, J. M. (2021). マウスのプライム編集により、組織特異的遺伝子発現を駆動する1塩基の特異性が明らかになった。ゲノム研究, 22(1), 1-21.
PDF版(英語)